Guide
Why VHP Sterilization
The complete reference for medical device manufacturers evaluating vaporized hydrogen peroxide sterilization — from mechanism to market clearance.
01
What Is VHP Sterilization?
Vaporized hydrogen peroxide (VHP) sterilization uses hydrogen peroxide in its gas phase to achieve microbial inactivation. The process operates at or near room temperature, leaves no toxic residue, and is compatible with a wide range of materials that cannot tolerate heat, radiation, or chemical sterilants.
VHP is a terminal sterilization method — meaning it achieves sterility assurance level (SAL) 10⁻⁶, the same assurance required of steam, ethylene oxide, and gamma irradiation for implantable medical devices. It is not a disinfection or surface decontamination process.
The hydrogen peroxide decomposes to water vapor and oxygen after the sterilization cycle. There are no toxic byproducts, no carcinogenic residues, and no aeration period required before product release.
SAL 10⁻⁶
Terminal sterility assurance — same standard as EtO and gamma irradiation
25–50°C
Operating temperature range — compatible with heat-sensitive materials
H₂O + O₂
Only decomposition products — zero toxic residue
02
How VHP Achieves Sterilization
VHP sterilization operates through gas-phase oxidation — a fundamentally different mechanism from irradiation, chemical alkylation (EtO), or thermal denaturation (steam).
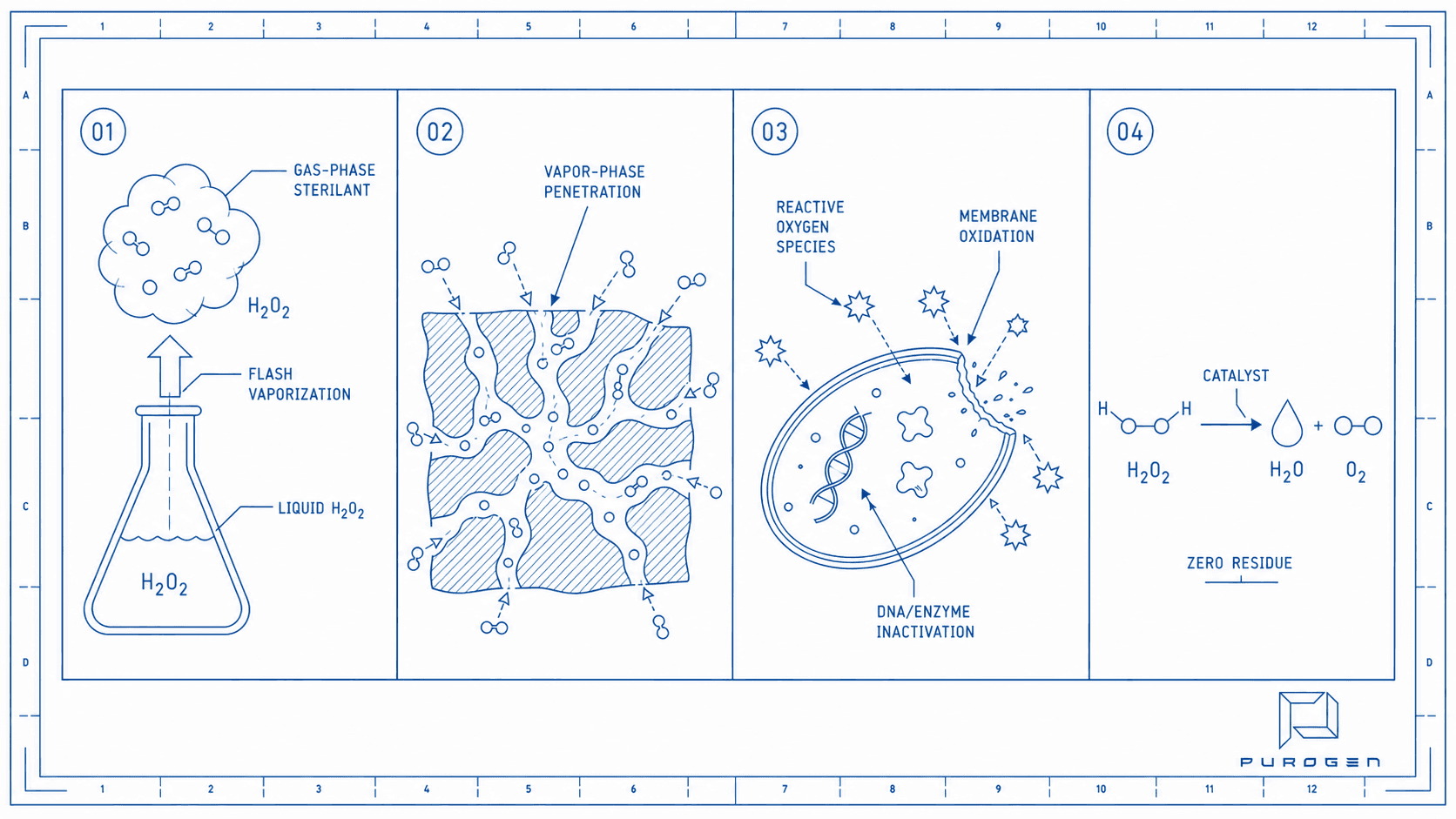
Generation
Liquid hydrogen peroxide is flash-vaporized into a gas-phase sterilant. Precise concentration control ensures reproducible dosing across every cycle.
Distribution
Vaporized H₂O₂ penetrates complex geometries, lumens, and porous materials. The vapor phase reaches surfaces that liquid and plasma methods cannot access.
Sterilization
Reactive oxygen species oxidize microbial cell membranes, DNA, and essential enzymes. Achieves SAL 10⁻⁶ — the gold standard for terminal sterilization.
Decomposition
H₂O₂ catalytically decomposes to water vapor and oxygen. Zero toxic residue. No aeration required. No environmental burden.
03
Material Compatibility
VHP's room-temperature oxidative mechanism is compatible with the broadest range of device materials of any established sterilization method. This is why VHP is the primary alternative for devices that cannot tolerate irradiation or heat.
Polymers
PEEK, PTFE, polyethylene, polypropylene, silicone, PVC, ABS, polycarbonate
Metals
Stainless steel, titanium, cobalt-chrome, aluminum alloys, nitinol
Electronics
Sensors, circuit boards, battery-powered devices, microprocessors
HEPA Filters
Filter media integrity preserved through low-temperature processing
Optics
Lenses, fiber optics, camera systems, light guides for endoscopic devices
Composites
Multi-material assemblies, coated surfaces, adhesive bonds, laminates
For device manufacturers with material sensitivity that precludes irradiation — due to polymer degradation, electronic component damage, or biological activity concerns — VHP is the only established alternative with a complete regulatory pathway.
04
Regulatory Framework: U.S. and International
VHP sterilization now operates within a fully defined regulatory and standards framework — across the United States, the European Union, and every market that references ISO standards.
Category A
As of January 8, 2024, the FDA classifies VHP as a Category A (Established) sterilization method — alongside steam, dry heat, EtO, and irradiation. Category A reduces the regulatory burden for demonstrating method suitability in 510(k) and PMA submissions.
Validation standard: ISO 22441
MDR 2017/745
Under EU MDR 2017/745, Notified Bodies assess sterilization processes against harmonized ISO standards. ISO 22441 provides VHP with the same standards-based CE marking pathway that EtO manufacturers use through ISO 11135 — without novel method justification.
Harmonized standard: ISO 22441
22441:2022
ISO 22441 is the international consensus standard for VHP sterilization of healthcare products. Referenced by the FDA, EU Notified Bodies, Health Canada, TGA (Australia), and regulatory authorities across Asia-Pacific.
One standard. Multiple market clearances.
FDA Sterilization Category Framework
Extensive published literature, validated biological indicators, established pathways. Steam, EtO, Radiation, VHP (Jan 2024).
Growing evidence base, no consensus standard. Higher regulatory burden. Ozone, Nitrogen Dioxide, Chlorine Dioxide.
Foundational research stage. No established pathway. Supercritical CO₂ (most device applications), other novel approaches.
05
VHP vs. Alternative Sterilization Methods
Five established modalities compared across the criteria that matter most to regulated device manufacturers.
| VHP | EtO | Gamma | Steam | E-beam | |
|---|---|---|---|---|---|
| Operating Temperature | Room temperature (25–50°C) | 37–63°C | Ambient | 121–134°C | Ambient |
| Toxic Residue | None — decomposes to H₂O + O₂ | Carcinogenic residues require aeration | None | None | None |
| Material Compatibility | Excellent — polymers, metals, electronics | Good for most materials | Degrades polymers and biologics | Limited — heat-sensitive materials excluded | Degrades some polymers |
| Biological Integrity | Preserved — osteoinductivity, growth factors intact | Generally preserved | Damaged — reduced mechanical properties | Damaged — protein denaturation | Partially damaged |
| Radiation Source | Not required | Not required | Cobalt-60 source required | Not required | Electron accelerator required |
| Aeration Time | None | 12–72 hours required | None | Drying cycle required | None |
| Regulatory Status | FDA Category A (Jan 2024), ISO 22441, EU MDR harmonized | Established — under EPA restriction | Established | Established | Established |
| Environmental Impact | Minimal — water and oxygen byproducts | Significant — EPA mandating 90% emission cuts | Radioactive source disposal | High energy consumption | Moderate energy consumption |
Operating Temperature
Toxic Residue
Material Compatibility
Biological Integrity
Radiation Source
Aeration Time
Regulatory Status
Environmental Impact
Within VHP
Not all VHP platforms perform within the same envelope.
The table above compares VHP to other sterilization categories. Within VHP itself, penetration depth, moisture operating range, and cycle architecture vary significantly between conventional systems and PuroGen's platform.
Three departures from conventional VHPPenetration
Driven into porous and dense substrates — not limited to surface contact and simple geometries.
Moisture Range
Up to ~20% RH vs. industry-typical ≤ 4% RH — a five-fold expansion of the operating envelope.
Cycle Authorship
PuroGen authors cycles specific to the customer's product. Customer validates. Customer's QMS owns the audit trail.
06
In-House vs. Contract Sterilization
The economics and operational implications of in-house VHP capability versus contract sterilization dependency.
In-House VHP Sterilization
Control over scheduling and cycle parameters
No per-unit contract fees — fixed capital amortization
Eliminates single-point-of-failure supply chain risk
Faster release cycles — no logistics to contract facility
Proprietary process knowledge stays internal
Scalable with volume growth
Contract Sterilization
No capital investment — pay-per-unit model
Validated process managed externally
Dependent on contract facility capacity and scheduling
Rising per-unit costs as EPA compliance investment increases
Supply chain vulnerability to facility disruptions
Limited ability to customize process for specific devices
Outsourced sterilization is a dependency. In-house sterilization is a capability. The economics of that distinction have shifted materially since EPA EtO mandates began compressing contract sterilization margins.
07
Validation Pathway
VHP sterilization validation follows the IQ/OQ/PQ framework defined in ISO 22441 — structured for FDA, EU, and international regulatory submissions.
Installation Qualification
Verification that the sterilization system is installed correctly and operates within specified parameters. Equipment calibration, utility connections, and documentation of as-built configuration.
Operational Qualification
Demonstration that the system operates consistently within defined process limits across the full range of operating conditions. Empty chamber studies, sensor mapping, and cycle parameter optimization.
Performance Qualification
Validation of the complete sterilization process under production conditions with representative product load configurations. Biological indicator challenges, sterility testing, and SAL 10⁻⁶ demonstration.
Multi-Market Documentation
ISO 22441 validation documentation is structured to support regulatory submissions across jurisdictions simultaneously. A single validation program — properly structured — produces documentation that satisfies FDA 510(k)/PMA requirements, EU MDR technical documentation requirements for Notified Body review, Health Canada device licensing, and TGA submissions. PuroGen structures validation documentation for multi-market use from the outset of the validation program.
08
Custom System Considerations
Off-the-shelf VHP systems are designed for generic applications. Medical device sterilization is not a generic application. Device geometry, material sensitivity, packaging configuration, and regulatory requirements vary by product — and each variation affects chamber design, cycle parameters, and validation scope.
PuroGen's SteriFlex platform is built for parametric customization: adjustable dwell times, reagent dosing, vacuum profiles, humidity control, and cycle sequencing. This means the sterilization process is engineered around your device — not the other way around. Custom chamber dimensions accommodate device form factors. Custom cycle parameters reflect your specific material profile.
The engagement model is flexible. License the SteriFlex platform for in-house sterilization with full validation support. Partner on a private-label basis for a system under your brand. Or engage in a strategic collaboration that leverages PuroGen's complete technology portfolio.